Below are the different processing steps your samples are subjected to after we receive them here at the core facility.
At TAMM, we are using an automated sample processing and tracking software platform in a laboratory information management system (Nautilus LIMS) to support high-throughput data generation and monitoring. Samples are tracked using barcode labels and non-personally identifiable tracking numbers. Genotyping steps include design of the assays, validation test run on our controls (DNAs for which SNP genotype is usually available from the HapMap Consortium), concordance check by re-genotyping a subset of customer’s samples, and the actual run on all material.
Genotyping of SNP
Design of assays: The assays are designed based on sequence information retrieved from the Ensembl database (www.ensembl.org), or in some cases sequence provided by the customer. The sequences are analyzed for proximal polymorphisms (which could affect the assay) as well as non-specific binding of the primers to the genome. This is performed using a design software package from Agena. Using the iPLEX chemistry, 36 SNPs can be analyzed in a single pool, i.e. you will obtain 36 genotypes per sample in a single reaction.
PCR: The first step in the analysis is a PCR reaction. The assays are set up in multiplexed reactions with primer pairs designed to amplify desired SNP loci.
PCR clean up: The PCR reaction is cleaned with an enzymatic reaction using SAP enzyme. This will remove the excess deoxynucleotides from the PCR reaction.
Extension reaction: Extension primers designed to anneal directly adjacent to the SNPs are added in a multiplexed single base pair extension (SBE) reaction with mass modified dideoxynucleotides.
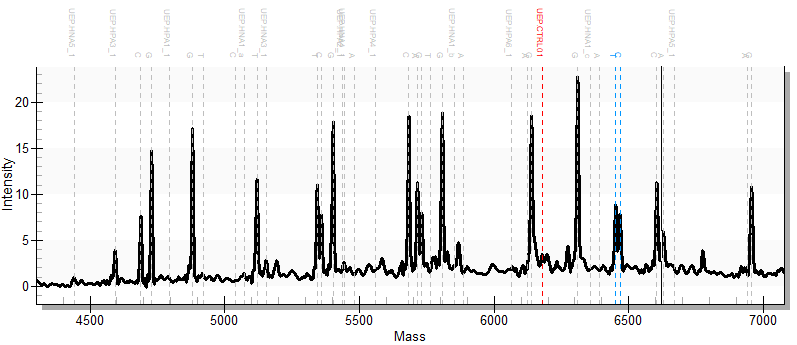
Fragment analysis: The extended primers are then subjected to MALDI-TOF analysis (Agena) on our Agena Massarray Analyzer.
Automated allele calling: We use Agena’s SpectroTyper software for automated allele calling, followed by thorough checking by two independent persons according to our SOPs (standard operating procedure). The analysis consistently gives genotyping calls with accuracy greater than 99%.
Validation: Marker polymorphism and suitability for genotyping is validated on a sample of human DNAs from the CEU population (US residents with northern and western European ancestry), which we have in house at MAF, and which have been genotyped by the Hapmap consortium (CEU panel). This enables us to perform concordance analysis of genotypes, when information on population frequencies of the SNP to be tested is publicly available. Positive and negative controls are included at this step and throughout the whole procedure.
Concordance: Genotyping of a number of samples is repeated to ensure reproducibility of the assay, and concordance analysis is thus carried out using this data. Prior to the analysis of the whole DNA panel, Validation and Concordance results are reported to the customer, together with Hardy-Weinberg equilibrium calculations.
Quality Control during large-scale genotyping: In each analysis unit (a 384 well plate) we include 24 controls consisting of 12 positive controls (the same CEU DNA repeated on all analysis units) and 12 negative controls. Family-related genotype data is checked for Mendelian inconsistencies using the PEDCHECK software (O’Connell and Weeks, 1998).
Genotyping results: The quality-controlled genotyping results are reported to the customer as genotypes, together with the quality scores for each assay performed. Our data output, usually summarized in an Excel file, is in a format compatible with existing genetic statistical software packages.